A few weeks ago, we quietly updated one component of our identity pricing options, making Tailscale usable with any OIDC-compliant SSO identity provider on any plan. It's a small change that reflects a bigger security principle.

|
Architechnosecurigeek. Tinkerer. General trouble maker.
|
This is the way it should be, and I'm happy to see one of my favorite services going in the right direction.
Arlington, VA

On March 7, 2024, tailscale.com was unavailable for approximately 90 minutes due to an expired TLS certificate. We were able to identify and address the issue quickly, and the downtime was mostly limited to our marketing materials and documentation; still, we would like to explain exactly what happened, what the impact was, and what steps we’ve taken to ensure it doesn’t happen again.
If it's not DNS, it's a PKI issue.
Arlington, VA

If necessity is the mother of invention, nostalgia must be its stepmother, or its aunt at the very least. The desire to recreate long-obsolete devices simply because they existed while we were growing up is a curious trait, but one that’s powerful enough to drive entire categories of hardware hacking — looking at you, retrocomputing buffs.
Hardware nostalgia isn’t all about 6502s and Z80s, though. Even more basic were the electronic toys of the 1970s, such as the Radio Shack 65-in-1 kit that [Tom Thoen] is currently recreating. The 65-in-1 was a breadboarding kit aimed at the budding electrical engineer, with components mounted to colorful cardboard by spring terminals. The included “lab manual” had circuits that could be quickly assembled using a handful of jumper wires. It was an endlessly fascinating toy that undoubtedly launched many careers, present company included.

While the passage of time may not have dulled [Tom]’s memories of his original 65-in-1, technology has marched on, meaning that certain allowances had to be made to create a modern version. He wisely eschews the cardboard for PCBs, one for each of the major component blocks provided in the original, and uses female header connectors in place of the springs. Component choice is tailored for the times; gone are the ferrite rod antenna and variable capacitor of the original, as well as the incandescent lamp, which is replaced by an LED that would have been a significant fraction of the kit’s $21.95 price back in 1976. There’s no BOM yet, so we can’t say for sure if any of the transistors are germanium, but it’s clear that there aren’t any of the old TO-1 cans. But dismay not, originalists, for the meter, relay, CdS photocell, and “solar battery” all made the final cut.
[Tom] has done some beautiful work here, with more to come. We imagine that 3D printing could be used to recreate some details like the original Morse key and speaker grille. We love the laser-engraved backing board, too, as it captures some of the charm of the original’s wooden box. This isn’t the only love for the “Science Fair” brand we’ve seen lately, either; the nostalgia seems to be contagious.
Arlington, VA
In 2017, we looked at how web bloat affects users with slow connections. Even in the U.S., many users didn't have broadband speeds, making much of the web difficult to use. It's still the case that many users don't have broadband speeds, both inside and outside of the U.S. and that much of the modern web isn't usable for people with slow internet, but the exponential increase in bandwidth (Nielsen suggests this is 50% per year for high-end connections) has outpaced web bloat for typical sites, making this less of a problem than it was in 2017, although it's still a serious problem for people with poor connections.
CPU performance for web apps hasn't scaled nearly as quickly as bandwidth so, while more of the web is becoming accessible to people with low-end connections, more of the web is becoming inaccessible to people with low-end devices even if they have high-end connections. For example, if I try browsing a "modern" Discourse-powered forum on a Tecno Spark 8C, it sometimes crashes the browser. Between crashes, on measuring the performance, the responsiveness is significantly worse than browsing a BBS with an 8 MHz 286 and a 1200 baud modem. On my 1Gbps home internet connection, the 2.6 MB compressed payload size "necessary" to load message titles is relatively light. The over-the-wire payload size has "only" increased by 1000x, which is dwarfed by the increase in internet speeds. But the opposite is true when it comes to CPU speeds — for web browsing and forum loading performance, the 8-core (2 1.6 GHz Cortex-A75 / 6 1.6 GHz Cortex-A55) CPU can't handle Discourse. The CPU is something like 100000x faster than our 286. Perhaps a 1000000x faster device would be sufficient.
For anyone not familiar with the Tecno Spark 8C, today, a new Tecno Spark 8C, a quick search indicates that one can be hand for USD 50-60 in Nigeria and perhaps USD 100-110 in India. As a fraction of median household income, that's substantially more than a current generation iPhone in the U.S. today.
By worldwide standards, the Tecno Spark 8C isn't even close to being a low-end device, so we'll also look at performance on an Itel P32, which is a lower end device (though still far from the lowest-end device people are using today). Additionally, we'll look at performance with an M3 Max Macbook (14-core), an M1 Pro Macbook (8-core), and the M3 Max set to 10x throttling in Chrome dev tools. In order to give these devices every advantage, we'll be on fairly high-speed internet (1Gbps, with a WiFi router that's benchmarked as having lower latency under load than most of its peers). We'll look at some blogging platforms and micro-blogging platforms (this blog, Substack, Medium, Ghost, Hugo, Tumblr, Mastodon, Twitter, Threads, Bluesky, Patreon), forum platforms (Discourse, Reddit, Quora, vBulletin, XenForo, phpBB, and myBB), and platforms commonly used by small businesses (Wix, Squarespace, Shopify, and WordPress again).
In the table below, every row represents a website and every non-label column is a metric. After the website name column, we have the compressed size transferred over the wire (wire) and the raw, uncompressed, size (raw). Then we have, for each device, Largest Contentful Paint* (LCP*) and CPU usage on the main thread (CPU). Google's docs explain LCP as
Largest Contentful Paint (LCP) measures when a user perceives that the largest content of a page is visible. The metric value for LCP represents the time duration between the user initiating the page load and the page rendering its primary content
LCP is a common optimization target because it's presented as one of the primary metrics in Google PageSpeed Insights, a "Core Web Vital" metric. There's an asterisk next to LCP as used in this document because, LCP as measured by Chrome is about painting a large fraction of the screen, as opposed to the definition above, which is about content. As sites have optimized for LCP, it's not uncommon to have a large paint (update) that's completely useless to the user, with the actual content of the page appearing well after the LCP. In cases where that happens, I've used the timestamp when useful content appears, not the LCP as defined by when a large but useless update occurs. The full details of the tests and why these metrics were chosen are discussed in an appendix.
Although CPU time isn't a "Core Web Vital", it's presented here because it's a simple metric that's highly correlated with my and other users' perception of usability on slow devices. See appendix for more detailed discussion on this. One reason CPU time works as a metric is that, if a page has great numbers for all other metrics but uses a ton of CPU time, the page is not going to be usable on a slow device. If it takes 100% CPU for 30 seconds, the page will be completely unusable for 30 seconds, and if it takes 50% CPU for 60 seconds, the page will be barely usable for 60 seconds, etc. Another reason it works is that, relative to commonly used metrics, it's hard to cheat on CPU time and make optimizations that significantly move the number without impacting user experience.
The color scheme in the table below is that, for sizes, more green = smaller / fast and more red = larger / slower. Extreme values are in black.
| Site | Size | M3 Max | M1 Pro | M3/10 | Tecno S8C | Itel P32 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wire | raw | LCP* | CPU | LCP* | CPU | LCP* | CPU | LCP* | CPU | LCP* | CPU | |
| danluu.com | 6kB | 18kB | 50ms | 20ms | 50ms | 30ms | 0.2s | 0.3s | 0.4s | 0.3s | 0.5s | 0.5s |
| HN | 11kB | 50kB | 0.1s | 30ms | 0.1s | 30ms | 0.3s | 0.3s | 0.5s | 0.5s | 0.7s | 0.6s |
| MyBB | 0.1MB | 0.3MB | 0.3s | 0.1s | 0.3s | 0.1s | 0.6s | 0.6s | 0.8s | 0.8s | 2.1s | 1.9s |
| phpBB | 0.4MB | 0.9MB | 0.3s | 0.1s | 0.4s | 0.1s | 0.7s | 1.1s | 1.7s | 1.5s | 4.1s | 3.9s |
| WordPress | 1.4MB | 1.7MB | 0.2s | 60ms | 0.2s | 80ms | 0.7s | 0.7s | 1s | 1.5s | 1.2s | 2.5s |
| WordPress (old) | 0.3MB | 1.0MB | 80ms | 70ms | 90ms | 90ms | 0.4s | 0.9s | 0.7s | 1.7s | 1.1s | 1.9s |
| XenForo | 0.3MB | 1.0MB | 0.4s | 0.1s | 0.6s | 0.2s | 1.4s | 1.5s | 1.5s | 1.8s | FAIL | FAIL |
| Ghost | 0.7MB | 2.4MB | 0.1s | 0.2s | 0.2s | 0.2s | 1.1s | 2.2s | 1s | 2.4s | 1.1s | 3.5s |
| vBulletin | 1.2MB | 3.4MB | 0.5s | 0.2s | 0.6s | 0.3s | 1.1s | 2.9s | 4.4s | 4.8s | 13s | 16s |
| Squarespace | 1.9MB | 7.1MB | 0.1s | 0.4s | 0.2s | 0.4s | 0.7s | 3.6s | 14s | 5.1s | 16s | 19s |
| Mastodon | 3.8MB | 5.3MB | 0.2s | 0.3s | 0.2s | 0.4s | 1.8s | 4.7s | 2.0s | 7.6s | FAIL | FAIL |
| Tumblr | 3.5MB | 7.1MB | 0.7s | 0.6s | 1.1s | 0.7s | 1.0s | 7.0s | 14s | 7.9s | 8.7s | 8.7s |
| Quora | 0.6MB | 4.9MB | 0.7s | 1.2s | 0.8s | 1.3s | 2.6s | 8.7s | FAIL | FAIL | 19s | 29s |
| Bluesky | 4.8MB | 10MB | 1.0s | 0.4s | 1.0s | 0.5s | 5.1s | 6.0s | 8.1s | 8.3s | FAIL | FAIL |
| Wix | 7.0MB | 21MB | 2.4s | 1.1s | 2.5s | 1.2s | 18s | 11s | 5.6s | 10s | FAIL | FAIL |
| Substack | 1.3MB | 4.3MB | 0.4s | 0.5s | 0.4s | 0.5s | 1.5s | 4.9s | 14s | 14s | FAIL | FAIL |
| Threads | 9.3MB | 13MB | 1.5s | 0.5s | 1.6s | 0.7s | 5.1s | 6.1s | 6.4s | 16s | 28s | 66s |
| 4.7MB | 11MB | 2.6s | 0.9s | 2.7s | 1.1s | 5.6s | 6.6s | 12s | 19s | 24s | 43s | |
| Shopify | 3.0MB | 5.5MB | 0.4s | 0.2s | 0.4s | 0.3s | 0.7s | 2.3s | 10s | 26s | FAIL | FAIL |
| Discourse | 2.6MB | 10MB | 1.1s | 0.5s | 1.5s | 0.6s | 6.5s | 5.9s | 15s | 26s | FAIL | FAIL |
| Patreon | 4.0MB | 13MB | 0.6s | 1.0s | 1.2s | 1.2s | 1.2s | 14s | 1.7s | 31s | 9.1s | 45s |
| Medium | 1.2MB | 3.3MB | 1.4s | 0.7s | 1.4s | 1s | 2s | 11s | 2.8s | 33s | 3.2s | 63s |
| 1.7MB | 5.4MB | 0.9s | 0.7s | 0.9s | 0.9s | 6.2s | 12s | 1.2s | ∞ | FAIL | FAIL | |
At a first glance, the table seems about right, in that the sites that feel slow unless you have a super fast device show up as slow in the table (as in, max(LCP*,CPU)) is high on lower-end devices). When I polled folks about what platforms they thought would be fastest and slowest on our slow devices (Mastodon, Twitter, Threads), they generally correctly predicted that Wordpress and Ghost and Wordpress would be faster than Substack and Medium, and that Discourse would be much slower than old PHP forums like phpBB, XenForo, and vBulletin. I also pulled Google PageSpeed Insights (PSI) scores for pages (not shown) and the correlation isn't as strong with those numbers because a handful of sites have managed to optimize their PSI scores without actually speeding up their pages for users.
If you've never used a low-end device like this, the general experience is that many sites are unusable on the device and loading anything resource intensive (an app or a huge website) can cause crashes. Doing something too intense in a resource intensive app can also cause crashes. While reviews note that you can run PUBG and other 3D games with decent performance on a Tecno Spark 8C, this doesn't mean that the device is fast enough to read posts on modern text-centric social media platforms or modern text-centric web forums. While 40fps is achievable in PUBG, we can easily see less than 0.4fps when scrolling on these sites.
We can see from the table how many of the sites are unusable if you have a slow device. All of the pages with 10s+ CPU are a fairly bad experience even after the page loads. Scrolling is very jerky, frequently dropping to a few frames per second and sometimes well below. When we tap on any link, the delay is so long that we can't be sure if our tap actually worked. If we tap again, we can get the dreaded situation where the first tap registers, which then causes the second tap to do the wrong thing, but if we wait, we often end up waiting too long because the original tap didn't actually register (or it registered, but not where we thought it did). Although MyBB doesn't serve up a mobile site and is penalized by Google for not having a mobile friendly page, it's actually much more usable on these slow mobiles than all but the fastest sites because scrolling and tapping actually work.
Another thing we can see is how much variance there is in the relative performance on different devices. For example, comparing an M3/10 and a Tecno Spark 8C, for danluu.com and Ghost, an M3/10 gives a halfway decent approximation of the Tecno Spark 8C (although danluu.com loads much too quickly), but the Tecno Spark 8C is about three times slower (CPU) for Medium, Substack, and Twitter, roughly four times slower for Reddit and Discourse, and over an order of magnitude faster for Shopify. For Wix, the CPU approximation is about accurate, but our `Tecno Spark 8C is more than 3 times slower on LCP*. It's great that Chrome lets you conveniently simulate a slower device from the convenience of your computer, but just enabling Chrome's CPU throttling (or using any combination of out-of-the-box options that are available) gives fairly different results than we get on many real devices. The full reasons for this are beyond the scope of the post; for the purposes of this post, it's sufficient to note that slow pages are often super-linearly slow as devices get slower and that slowness on one page doesn't strongly predict slowness on another page.
If take a site-centric view instead of a device-centric view, another way to look at it is that sites like Discourse, Medium, and Reddit, don't use all that much CPU on our fast M3 and M1 computers, but they're among the slowest on our Tecno Spark 8C (Reddit's CPU is shown as ∞ because, no matter how long we wait with no interaction, Reddit uses ~90% CPU). Discourse also sometimes crashed the browser after interacting a bit or just waiting a while. For example, one time, the browser crashed after loading Discourse, scrolling twice, and then leaving the device still for a minute or two. For consistency's sake, this wasn't marked as FAIL in the table since the page did load but, realistically, having a page is so resource intensive that the browser crashes is a significantly worse user experience than any of the FAIL cases in the table. When we looked at how web bloat impacts users with slow connections, we found that much of the web was unusable for people with slow connections and slow devices are no different.
Another pattern we can see is how the older sites are, in general, faster than the newer ones, with sites that (visually) look like they haven't been updated in a decade or two tending to be among the fastest. For example, MyBB, the least modernized and oldest looking forum is 3.6x / 5x faster (LCP* / CPU) than Discourse on the M3, but on the Tecno Spark 8C, the difference is 19x / 33x and, given the overall scaling, it seems safe to guess that the difference would be even larger on the Itel P32 if Discourse worked on such a cheap device.
Another example is Wordpress (old) vs. newer, trendier, blogging platforms like Medium and Substack. Wordpress (old) is is 17.5x / 10x faster (LCP* / CPU) than Medium and 5x / 7x faster (LCP* / CPU) faster than Substack on our M3 Max, and 4x / 19x and 20x / 8x faster, respectively, on our Tecno Spark 8C. Ghost is a notable exception to this, being a modern platform (launched a year after Medium) that's competitive with older platforms (modern Wordpress is also arguably an exception, but many folks would probably still consider that to be an old platform).
Sites that use modern techniques like partially loading the page and then dynamically loading the rest of it, such as Discourse, Reddit, and Substack, tend to be less usable than the scores in the table indicate. Although, in principle, you could build such a site in a simple way that works well with cheap devices but, in practice sites that use dynamic loading tend to be complex enough that the sites are extremely janky on low-end devices. It's generally difficult or impossible to scroll a predictable distance, which means that users will sometimes accidentally trigger more loading by scrolling too far, causing the page to lock up. Many pages actually remove the parts of the page you scrolled past as you scroll; all such pages are essentially unusable. Other basic web features, like page search, also generally stop working. Pages with this kind of dynamic loading can't rely on the simple and fast ctrl/command+F search and have to build their own search. How well this works varies (this used to work quite well in Google docs, but for the past few months or maybe a year, it takes so long to load that I have to deliberately wait after opening a doc to avoid triggering the browser's useless built in search; Discourse search has never really worked on slow devices or even not very fast but not particular slow devices).
In principle, these modern pages that burn a ton of CPU when loading could be doing pre-work that means that later interactions on the page are faster and cheaper than on the pages that do less up-front work (this is a common argument in favor of these kinds of pages), but that's not the case for pages tested, which are slower to load initially, slower on subsequent loads, and slower after they've loaded.
To understand why the theoretical idea that doing all this work up-front doesn't generally result in a faster experience later, this exchange between a distinguished engineer at Google and one of the founders of Discourse (and CEO at the time) is illustrative, in a discussion where the founder of Discourse says that you should test mobile sites on laptops with throttled bandwidth but not throttled CPU:
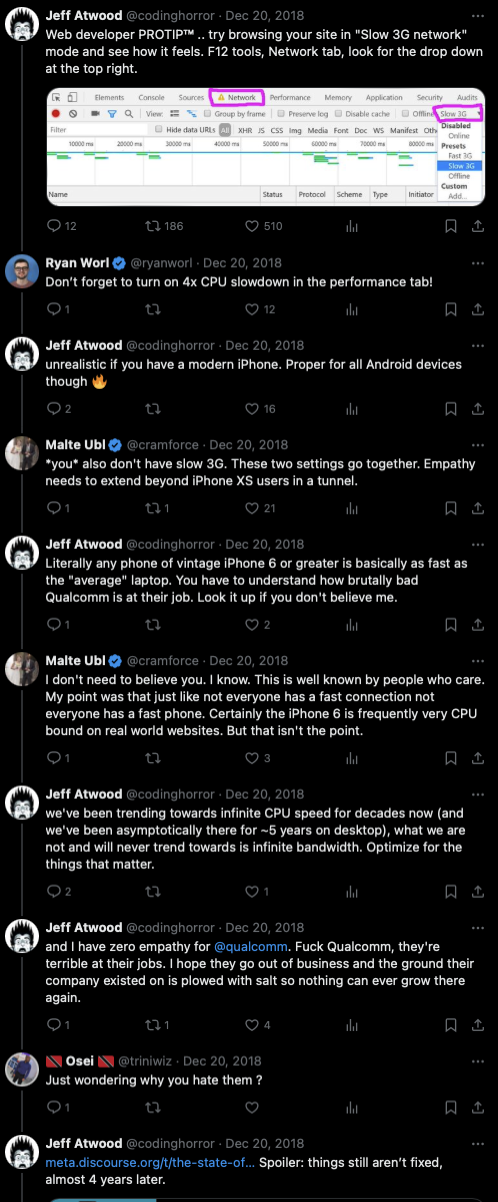{kind=link}
- Google: *you* also don't have slow 3G. These two settings go together. Empathy needs to extend beyond iPhone XS users in a tunnel.
- Discourse: Literally any phone of vintage iPhone 6 or greater is basically as fast as the "average" laptop. You have to understand how brutally bad Qualcomm is at their job. Look it up if you don't believe me.
- Google: I don't need to believe you. I know. This is well known by people who care. My point was that just like not everyone has a fast connection not everyone has a fast phone. Certainly the iPhone 6 is frequently very CPU bound on real world websites. But that isn't the point.
- Discourse: we've been trending towards infinite CPU speed for decades now (and we've been asymptotically there for ~5 years on desktop), what we are not and will never trend towards is infinite bandwidth. Optimize for the things that matter. and I have zero empathy for @qualcomm. Fuck Qualcomm, they're terrible at their jobs. I hope they go out of business and the ground their company existed on is plowed with salt so nothing can ever grow there again.
- Google: Mobile devices are not at all bandwidth constraint in most circumstances. They are latency constraint. Even the latest iPhone is CPU constraint before it is bandwidth constraint. If you do well on 4x slow down on a MBP things are pretty alright
- ...
- Google: Are 100% of users on iOS?
- Discourse: The influential users who spend money tend to be, I’ll tell you that ... Pointless to worry about cpu, it is effectively infinite already on iOS, and even with Qualcomm’s incompetence, will be within 4 more years on their embarrassing SoCs as well
When someone asks the founder of Discourse, "just wondering why you hate them", he responds with a link that cites the Kraken and Octane benchmarks from this Anandtech review, which have the Qualcomm chip at 74% and 85% of the performance of the then-current Apple chip, respectively.
The founder and then-CEO of Discourse considers Qualcomm's mobile performance embarrassing and finds this so offensive that he thinks Qualcomm engineers should all lose their jobs for delivering 74% to 85% of the performance of Apple. Apple has what I consider to be an all-time great performance team. Reasonable people could disagree on that, but one has to at least think of them as a world-class team. So, producing a product with 74% to 85% of an all-time-great team is considered an embarrassment worthy of losing your job.
There are two attitudes on display here which I see in a lot of software folks. First, that CPU speed is infinite and one shouldn't worry about CPU optimization. And second, that gigantic speedups from hardware should be expected and the only reason hardware engineers wouldn't achieve them is due to spectacular incompetence, so the slow software should be blamed on hardware engineers, not software engineers. Donald Knuth expressed a similar sentiment in
I might as well flame a bit about my personal unhappiness with the current trend toward multicore architecture. To me, it looks more or less like the hardware designers have run out of ideas, and that they’re trying to pass the blame for the future demise of Moore’s Law to the software writers by giving us machines that work faster only on a few key benchmarks! I won’t be surprised at all if the whole multiithreading idea turns out to be a flop, worse than the "Itanium" approach that was supposed to be so terrific—until it turned out that the wished-for compilers were basically impossible to write. Let me put it this way: During the past 50 years, I’ve written well over a thousand programs, many of which have substantial size. I can’t think of even five of those programs that would have been enhanced noticeably by parallelism or multithreading. Surely, for example, multiple processors are no help to TeX ... I know that important applications for parallelism exist—rendering graphics, breaking codes, scanning images, simulating physical and biological processes, etc. But all these applications require dedicated code and special-purpose techniques, which will need to be changed substantially every few years. Even if I knew enough about such methods to write about them in TAOCP, my time would be largely wasted, because soon there would be little reason for anybody to read those parts ... The machine I use today has dual processors. I get to use them both only when I’m running two independent jobs at the same time; that’s nice, but it happens only a few minutes every week.
In the case of Discourse, a hardware engineer is an embarrassment not deserving of a job if they can't hit 90% of the performance of an all-time-great performance team but, as a software engineer, delivering 3% the performance of a non-highly-optimized application like MyBB is no problem. In Knuth's case, hardware engineers gave programmers a 100x performance increase every decade for decades with little to no work on the part of programmers. The moment this slowed down and programmers had to adapt to take advantage of new hardware, hardware engineers were "all out of ideas", but learning a few "new" (1970s and 1980s era) ideas to take advantage of current hardware would be a waste of time. And we've previously discussed Alan Kay's claim that hardware engineers are "unsophisticated" and "uneducated" and aren't doing "real engineering" and how we'd get a 1000x speedup if we listened to Alan Kay's "sophisticated" ideas.
It's fairly common for programmers to expect that hardware will solve all their problems, and then, when that doesn't happen, pass the issue onto the user, explaining why the programmer needn't do anything to help the user. A question one might ask is how much performance improvement programmers have given us. There are cases of algorithmic improvements that result in massive speedups but, as we noted above, Discourse, the fastest growing forum software today, seems to have given us an approximately 1000000x slowdown in performance.
Another common attitude on display above is the idea that users who aren't wealthy don't matter. When asked if 100% of users are on iOS, the founder of Discourse says "The influential users who spend money tend to be, I’ll tell you that". We see the same attitude all over comments on Tonsky's JavaScript Bloat post, with people expressing cocktail-party sentiments like "Phone apps are hundreds of megs, why are we obsessing over web apps that are a few megs? Starving children in Africa can download Android apps but not web apps? Come on" and "surely no user of gitlab would be poor enough to have a slow device, let's be serious" (paraphrased for length).
But when we look at the size of apps that are downloaded in Africa, we see that people who aren't on high-end devices use apps like Facebook Lite (a couple megs) and commonly use apps that are a single digit to low double digit number of megabytes. There are multiple reasons app makers care about their app size. One is just the total storage available on the phone; if you watch real users install apps, they often have to delete and uninstall things to put a new app on, so the smaller size is both easier to to install and has a lower chance of being uninstalled when the user is looking for more space. Another is that, if you look at data on app size and usage (I don't know of any public data on this; please pass it along if you have something public I can reference), when large apps increase the size and memory usage, they get more crashes, which drives down user retention, growth, and engagement and, conversely, when they optimize their size and memory usage, they get fewer crashes and better user retention, growth, and engagement.
Alex Russell points out that iOS has 7% market share in India (a 1.4B person market) and 6% market share in Latin America (a 600M person market). Although the founder of Discourse says that these aren't "influential users" who matter, these are still real human beings. Alex further points out that, according to Windows telemetry, which covers the vast majority of desktop users, most laptop/desktop users are on low-end machines which are likely slower than a modern iPhone.
On the bit about no programmers having slow devices, I know plenty of people who are using hand-me-down devices that are old and slow. Many of them aren't even really poor; they just don't see why (for example) their kid needs a super fast device, and they don't understand how much of the modern web works poorly on slow devices. After all, the "slow" device can play 3d games and (with the right OS) compile codebases like Linux or Chromium, so why shouldn't the device be able to interact with a site like gitlab?
Contrary to the claim from the founder of Discourse that, within years, every Android user will be on some kind of super fast Android device, it's been six years since his comment and it's going to be at least a decade before almost everyone in the world who's using a phone has a high-speed device and this could easily take two decades or more. If you look up marketshare stats for Discourse, it's extremely successful; it appears to be the fastest growing forum software in the world by a large margin. The impact of having the fastest growing forum software in the world created by an organization whose then-leader was willing to state that he doesn't really care about users who aren't "influential users who spend money", who don't have access to "infinite CPU speed", is that a lot of forums are now inaccessible to people who don't have enough wealth to buy a device with effectively infinite CPU.
If the founder of Discourse were an anomaly, this wouldn't be too much of a problem, but he's just verbalizing the implicit assumptions a lot of programmers have, which is why we see that so many modern websites are unusable if you buy the income-adjusted equivalent of a new, current generation, iPhone in a low-income country.
Thanks to Yossi Kreinen, Fabian Giesen, John O'Nolan, Joseph Scott, Loren McIntyre, Daniel Filan, @acidshill, Alex Russell, Chris Adams, Tobias Marschner, Matt Stuchlik, @gekitsu@toot.cat, Justin Blank, Andy Kelley, Julian Lam, and David Turner for comments/corrections/discussion.
Appendix: gaming LCP
We noted above that we used LCP* and not LCP. This is because LCP basically measures when the largest change happens. When this metric was not deliberately gamed in ways that don't benefit the user, this was a great metric, but this metric has become less representative of the actual user experience as more people have gamed it. In the less blatant cases, people do small optimizations that improve LCP but barely improve or don't improve the actual user experience.
In the more blatant cases, developers will deliberately flash a very large change on the page as soon as possible, generally a loading screen that has no value to the user (actually negative value because doing this increases the total amount of work done and the total time it takes to load the page) and then they carefully avoid making any change large enough that any later change would get marked as the LCP.
For the same reason that VW didn't publicly discuss how it was gaming its emissions numbers, developers tend to shy away from discussing this kind of LCP optimization in public. An exception to this is Discourse, where they publicly announced this kind of LCP optimization, with comments from their devs and the then-CTO (now CEO), noting that their new "Discourse Splash" feature hugely reduced LCP for sites after they deployed it. And then developers ask why their LCP is high, the standard advice from Discourse developers is to keep elements smaller than the "Discourse Splash", so that the LCP timestamp is computed from this useless element that's thrown up to optimize LCP, as opposed to having the timestamp be computed from any actual element that's relevant to the user. Here's a typical, official, comment from Discourse
If your banner is larger than the element we use for the "Introducing Discourse Splash - A visual preloader displayed while site assets load" you gonna have a bad time for LCP.
The official response from Discourse is that you should make sure that your content doesn't trigger the LCP measurement and that, instead, our loading animation timestamp is what's used to compute `LCP.
The sites with the most extreme ratio of LCP of useful content vs. Chrome's measured LCP were:
- Wix
M3:6M1:12Tecno Spark 8C:3Itel P32:N/A(FAIL)
- Discourse:
M3:10M1:12Tecno Spark 8C:4Itel P32:N/A(FAIL)
Although we haven't discussed the gaming of other metrics, it appears that some websites also game other metrics and "optimize" them even when this has no benefit to users.
Appendix: the selfish argument for optimizing sites
This will depend on the scale of the site as well as its performance, but when I've looked at this data for large companies I've worked for, improving site and app performance is worth a mind boggling amount of money. It's measurable in A/B tests and it's also among the interventions that has, in long-term holdbacks, a relatively large impact on growth and retention (many interventions test well but don't look as good long term, whereas performance improvements tend to look better long term).
Of course you can see this from the direct numbers, but you can also implicitly see this in a lot of ways when looking at the data. One angle is that (just for example), at Twitter, user-observed p99 latency was about 60s in India as well as a number of African countries (even excluding relatively wealthy ones like Egypt and South Africa) and also about 60s in the United States. Of course, across the entire population, people have faster devices and connections in the United States, but in every country, there are enough users that have slow devices or connections that the limiting factor is really user patience and not the underlying population-level distribution of devices and connections. Even if you don't care about users in Nigeria or India and only care about U.S. ad revenue, improving performance for low-end devices and connections has enough of impact that we could easily see the impact in global as well as U.S. revenue in A/B tests, especially in long-term holdbacks. And you also see the impact among users who have fast devices since a change that improves the latency for a user with a "low-end" device from 60s to 50s might improve the latency for a user with a high-end device from 5s to 4.5s, which has an impact on revenue, growth, and retention numbers as well.
For a variety of reasons that are beyond the scope of this doc, this kind of boring, quantifiable, growth and revenue driving work has been difficult to get funded at most large companies I've worked for relative to flash product work that ends up showing little to no impact in long-term holdbacks.
Appendix: designing for low performance devices
When using slow devices or any device with low bandwidth and/or poor connectivity, the best experiences, by far, are generally the ones that load a lot of content at once into a static page. If the images have proper width and height attributes and alt text, that's very helpful. Progressive images (as in progressive jpeg) isn't particularly helpful.
On a slow device with high bandwidth, any lightweight, static, page works well, and lightweight dynamic pages can work well if designed for performance. Heavy, dynamic, pages are doomed unless the page weight doesn't cause the page to be complex.
With low bandwidth and/or poor connectivity, lightweight pages are fine. With heavy pages, the best experience I've had is when I trigger a page load, go do something else, and then come back when it's done (or at least the HTML and CSS are done). I can then open each link I might want to read in a new tab, and then do something else while I wait for those to load.
A lot of the optimizations that modern websites do, such as partial loading that causes more loading when you scroll down the page, and the concomitant hijacking of search (because the browser's built in search is useless if the page isn't fully loaded) causes the interaction model that works to stop working and makes pages very painful to interact with.
Just for example, a number of people have noted that Substack performs poorly for them because it does partial page loads. Here's a video by @acidshill of what it looks like to load a Substack article and then scroll on an iPhone 8, where the post has a fairly fast LCP, but if you want to scroll past the header, you have to wait 6s for the next page to load, and then on scrolling again, you have to wait maybe another 1s to 2s:
As an example of the opposite approach, I tried loading some fairly large plain HTML pages, such as https://danluu.com/diseconomies-scale/ (0.1 MB wire / 0.4 MB raw) and https://danluu.com/threads-faq/ (0.4 MB wire / 1.1 MB raw) and these were still quite usable for me even on slow devices. 1.1 MB seems to be larger than optimal and breaking that into a few different pages would be better on a low-end devices, but a single page with 1.1 MB of text works much better than most modern sites on a slow device. While you can get into trouble with HTML pages that are so large that browsers can't really handle them, for pages with a normal amount of content, it generally isn't until you have complex CSS payloads or JS that the pages start causing problems for slow devices. Below, we test pages that are relatively simple, some of which have a fair amount of media (14 MB in one case) and find that these pages work ok, as long as they stay simple.
Chris Adams has also noted that blind users, using screen readers, often report that dynamic loading makes the experience much worse for them. Like dynamic loading to improve performance, while this can be done well, it's often either done badly or bundled with so much other complexity that the result is worse than a simple page.
@Qingcharles noted another accessibility issue — the (prison) parolees he works with are given "lifeline" phones, which are often very low end devices. From a quick search, in 2024, some people will get an iPhone 6 or an iPhone 8, but there are also plenty of devices that are lower end than an Itel P32, let alone a Tecno Spark 8C. They also get plans with highly limited data, and then when they run out, some people "can't fill out any forms for jobs, welfare, or navigate anywhere with Maps".
For sites that do up-front work and actually give you a decent experience on low end devices, Andy Kelley pointed out an example of a site that does up front work that seems to work ok on a slow device (although it would struggle on a very slow connection), the Zig standard library documentation:
I made the controversial decision to have it fetch all the source code up front and then do all the content rendering locally. In theory, this is CPU intensive but in practice... even those old phones have really fast CPUs!
On the Tecno Spark 8C, this uses 4.7s of CPU and, afterwards, is fairly responsive (relative to the device — of course an iPhone responds much more quickly. Taps cause links to load fairly quickly and scrolling also works fine (it's a little jerky, but almost nothing is really smooth on this device). This seems like the kind of thing people are referring to when they say that you can get better performance if you ship a heavy payload, but there aren't many examples of that which actually improve performance on low-end devices.
Appendix: articles on web performance issues
- 2015: Maciej Cegłowski: The Website Obesity Crisis
- Size:
1.0 MB/1.1 MB Tecno Spark 8C:0.9s/1.4s- Scrolling a bit jerky, images take a little bit of time to appear if scrolling very quickly (jumping halfway down page from top), but delay is below what almost any user would perceive when scrolling a normal distance.
- Size:
- 2015: Nate Berkopec: Page Weight Doesn't Matter
- Size:
80 kB/0.2 MB Tecno Spark 8C:0.8s/0.7s- Does lazy loading, page downloads
650 kB/1.8 MBif you scroll through the entire page, but scrolling is only a little jerky and the lazy loading doesn't cause delays. Probably the only page I've tried that does lazy loading in a way that makes the experience better and not worse on a slow device; I didn't test on a slow connection, where this would still make the experience worse.
- Does lazy loading, page downloads
Itel P32:1.1s/1s- Scrolling basically unusable; scroll extremely jerky and moves a random distance, often takes over
1sfor text to render when scrolling to new text; can be much worse with images that are lazy loaded. Even though this is the best implementation of lazy loading I've seen in the wild, theItel P32still can't handle it.
- Scrolling basically unusable; scroll extremely jerky and moves a random distance, often takes over
- Size:
- 2017: Dan Luu: How web bloat impacts users with slow connections
- Size:
14 kB/57 kB Tecno Spark 8C:0.5s/0.3s- Scrolling and interaction work fine.
Itel P32:0.7s/0.5 s
- Size:
- 2017-2024+: Alex Russell: The Performance Inequality Gap (series)
- Size:
82 kB/0.1 MB Tecno Spark 8C:0.5s/0.4s- Scrolling and interaction work fine.
Itel P32:0.7s/0.4s- Scrolling and interaction work fine.
- Size:
- 2024: Nikita Prokopov (Tonsky): JavaScript Bloat in 2024
- Size:
14 MB/14 MB Tecno Spark 8C:0.8s/1.9s- When scrolling, it takes a while for images to show up (500ms or so) and the scrolling isn't smooth, but it's not jerky enough that it's difficult to scroll to the right place.
Itel P32:2.5s/3s- Scrolling isn't smooth. Scrolling accurately is a bit difficult, but can generally scroll to where you want if very careful. Generally takes a bit more than
1sfor new content to appear when you scroll a significant distance.
- Scrolling isn't smooth. Scrolling accurately is a bit difficult, but can generally scroll to where you want if very careful. Generally takes a bit more than
- Size:
- 2024: Dan Luu: This post
- Size:
25 kB/74 kB Tecno Spark 8C:0.6s/0.5s- Scrolling and interaction work fine.
Itel P32:1.3s/1.1s- Scrolling and interaction work fine, although I had to make a change for this to be the case — this doc originally had an embedded video, which the
Itel P32couldn't really handle.- Note that, while these numbers are worse than the numbers for "Page Weight Doesn't Matter", this page is usable after load, which that other page isn't beacuse it execute some kind of lazy loading that's too complex for this phone to handle in a reasonable timeframe.
- Note that, while these numbers are worse than the numbers for "Page Weight Doesn't Matter", this page is usable after load, which that other page isn't beacuse it execute some kind of lazy loading that's too complex for this phone to handle in a reasonable timeframe.
- Scrolling and interaction work fine, although I had to make a change for this to be the case — this doc originally had an embedded video, which the
- Size:
Appendix: empathy for non-rich users
Something I've observed over time, as programming has become more prestigious and more lucrative, is that people have tended to come from wealthier backgrounds and have less exposure to people with different income levels. An example we've discussed before, is at a well-known, prestigious, startup that has a very left-leaning employee base, where everyone got rich, on a discussion about the covid stimulus checks, in a slack discussion, a well meaning progressive employee said that it was pointless because people would just use their stimulus checks to buy stock. This person had, apparently, never talked to any middle-class (let alone poor) person about where their money goes or looked at the data on who owns equity. And that's just looking at American wealth. When we look at world-wide wealth, the general level of understanding is much lower. People seem to really underestimate the dynamic range in wealth and income across the world. From having talked to quite a few people about this, a lot of people seem to have mental buckets for "poor by American standards" (buys stock with stimulus checks) and "poor by worldwide standards" (maybe doesn't even buy stock), but the range of poverty in the world dwarfs the range of poverty in America to an extent that not many wealthy programmers seem to realize.
Just for example, in this discussion how lucky I was (in terms of financial opportunities) that my parents made it to America, someone mentioned that it's not that big a deal because they had great financial opportunities in Poland. For one thing, with respect to the topic of the discussion, the probability that someone will end up with a high-paying programming job (senior staff eng at a high-paying tech company) or equivalent, I suspect that, when I was born, being born poor in the U.S. gives you better odds than being fairly well off in Poland, but I could believe the other case as well if presented with data. But if we're comparing Poland v. U.S. to Vietnam v. U.S., if I spend 15 seconds looking up rough wealth numbers for these countries in the year I was born, the GDP/capita ratio of U.S. : Poland was ~8:1, whereas it was ~50 : 1 for Poland : Vietnam. The difference in wealth between Poland and Vietnam was roughly the square of the difference between the U.S. and Poland, so Poland to Vietnam is roughly equivalent to Poland vs. some hypothetical country that's richer than the U.S. by the amount that the U.S. is richer than Poland. These aren't even remotely comparable, but a lot of people seem to have this mental model that there's "rich countries" and "not rich countries" and "not rich countries" are all roughly in the same bucket. GDP/capita isn't ideal, but it's easier to find than percentile income statistics; the quick search I did also turned up that annual income in Vietnam then was something like $200-$300 a year. Vietnam was also going through the tail end of a famine whose impacts are a bit difficult to determine because statistics here seem to be gamed, but if you believe the mortality rate statistics, the famine caused total overall mortality rate to jump to double the normal baseline1.
Of course, at the time, the median person in a low-income country wouldn't have had a computer, let alone internet access. But, today it's fairly common for people in low-income countries to have devices. Many people either don't seem to realize this or don't understand what sorts of devices a lot of these folks use.
Appendix: comments from Fabian Giesen
On the Discourse founder's comments on iOS vs. Android marketshare, Fabian notes
In the US, according to the most recent data I could find (for 2023), iPhones have around 60% marketshare. In the EU, it's around 33%. This has knock-on effects. Not only do iOS users skew towards the wealthier end, they also skew towards the US.
There's some secondary effects from this too. For example, in the US, iMessage is very popular for group chats etc. and infamous for interoperating very poorly with Android devices in a way that makes the experience for Android users very annoying (almost certainly intentionally so).
In the EU, not least because Android is so much more prominent, iMessage is way less popular and anecdotally, even iPhone users among my acquaintances who would probably use iMessage in the US tend to use WhatsApp instead.
Point being, globally speaking, recent iOS + fast Internet is even more skewed towards a particular demographic than many app devs in the US seem to be aware.
And on the comment about mobile app vs. web app sizes, Fabian said:
One more note from experience: apps you install when you install them, and generally have some opportunity to hold off on updates while you're on a slow or metered connection (or just don't have data at all).
Back when I originally got my US phone, I had no US credit history and thus had to use prepaid plans. I still do because it's fine for what I actually use my phone for most of the time, but it does mean that when I travel to Germany once a year, I don't get data roaming at all. (Also, phone calls in Germany cost me $1.50 apiece, even though T-Mobile is the biggest mobile provider in Germany - though, of course, not T-Mobile US.)
Point being, I do get access to free and fast Wi-Fi at T-Mobile hotspots (e.g. major train stations, airports etc.) and on inter-city trains that have them, but I effectively don't have any data plan when in Germany at all.
This is completely fine with mobile phone apps that work offline and sync their data when they have a connection. But web apps are unusable while I'm not near a public Wi-Fi.
Likewise I'm fine sending an email over a slow metered connection via the Gmail app, but I for sure wouldn't use any web-mail client that needs to download a few MBs worth of zipped JS to do anything on a metered connection.
At least with native app downloads, I can prepare in advance and download them while I'm somewhere with good internet!
Another comment from Fabian (this time paraphrased since this was from a conversation), is that people will often justify being quantitatively hugely slower because there's a qualitative reason something should be slow. One example he gave was that screens often take a long time to sync their connection and this is justified because there are operations that have to be done that take time. For a long time, these operations would often take seconds. Recently, a lot of displays sync much more quickly because Nvidia specifies how long this can take for something to be "G-Sync" certified, so display makers actually do this in a reasonable amount of time now. While it's true that there are operations that have to be done that take time, there's no fundamental reason they should take as much time as they often used to. Another example he gave was on how someone was justifying how long it took to read thousands of files because the operation required a lot of syscalls and "syscalls are slow", which is a qualitatively true statement, but if you look at the actual cost of a syscall, in the case under discussion, the cost of a syscall was many orders of magnitude from being costly enough to be a reasonable explanation for why it took so long to read thousands of files.
On this topic, when people point out that a modern website is slow, someone will generally respond with the qualitative defense that the modern website has these great features, which the older website is lacking. And while it's true that (for example) Discourse has features that MyBB doesn't, it's hard to argue that its feature set justifies being 33x slower.
Appendix: experimental details
With the exception of danluu.com and, arguably, HN, for each site, I tried to find the "most default" experience. For example, for WordPress, this meant a demo blog with the current default theme, twentytwentyfour. In some cases, this may not be the most likely thing someone uses today, e.g., for Shopify, I looked at the first thing that theme they give you when you browse their themes, but I didn't attempt to find theme data to see what the most commonly used theme is. For this post, I wanted to do all of the data collection and analysis as a short project, something that takes less than a day, so there were a number of shortcuts like this, which will be described below. I don't think it's wrong to use the first-presented Shopify theme in a decent fraction of users will probably use the first-presente theme, but that is, of course, less representative than grabbing whatever the most common theme is and then also testing many different sites that use that theme to see how real-world performance varies when people modify the theme for their own use. If I worked for Shopify or wanted to do competitive analysis on behalf of a competitor, I would do that, but for a one-day project on how large websites impact users on low-end devices, the performance of Shopify demonstrated here seems ok. I actually did the initial work for this around when I ran these polls, back in February; I just didn't have time to really write this stuff up for a month.
For the tests on laptops, I tried to have the laptop at ~60% battery, not plugged in, and the laptop was idle for enough time to return to thermal equilibrium in a room at 20°C, so pages shouldn't be impacted by prior page loads or other prior work that was happening on the machine.
For the mobile tests, the phones were at ~100% charge and plugged in, and also previously at 100% charge so the phones didn't have any heating effect you can get from rapidly charging. As noted above, these tests were formed with 1Gbps WiFi. No other apps were running, the browser had no other tabs open, and the only apps that were installed on the device, so no additional background tasks should've been running other than whatever users are normally subject to by the device by default. A real user with the same device is going to see worse performance than we measured here in almost every circumstance except if running Chrome Dev Tools on a phone significantly degrades performance. I noticed that, on the Itel P32, scrolling was somewhat jerkier with Dev Tools running than when running normally but, since this was a one-day project, I didn't attempt to quantify this and if it impacts some sites much more than others. In absolute terms, the overhead can't be all that large because the fastest sites are still fairly fast with Dev Tools running, but if there's some kind of overhead that's super-linear in the amount of work the site does (possibly indirectly, if it causes some kind of resource exhaustion), then that could be a problem in measurements of some sites.
Sizes were all measured on mobile, so in cases where different assets are loaded on mobile vs. desktop, the we measured the mobile asset sizes. CPU was measured as CPU time on the main thread (I did also record time on other threads for sites that used other threads, but didn't use this number; if CPU were a metric people wanted to game, time on other threads would have to be accounted for to prevent sites from trying to offload as much work as possible to other threads, but this isn't currently an issue and time on main thread is more directly correlated to usability than sum of time across all threads, and the metric that would work for gaming is less legible with no upside for now).
For WiFi speeds, speed tests had the following numbers:
M3 Max- Netflix (fast.com)
- Download:
850 Mbps - Upload:
840 Mbps - Latency (unloaded / loaded):
3ms/8ms
- Download:
- Okta
- Download:
900 Mbps - Upload:
840 Mbps - Latency (unloaded / download / upload):
3ms/8ms/13ms
- Download:
- Netflix (fast.com)
Tecno Spark 8C- Netflix (fast.com)
- Download:
390 Mbps - Upload:
210 Mbps - Latency (unloaded / loaded):
2ms/30ms
- Download:
- Okta
- Okta web app fails, can't see results
- Netflix (fast.com)
Itel P32- Netflix
- Download:
44 Mbps - Upload: test fails to work (sends one chunk of data and then hangs, sending no more data)
- Latency (unloaded / loaded):
4ms/400ms
- Download:
- Okta
- Download:
45 Mbps - Upload: test fails to work
- Latency: test fails to display latency
- Download:
- Netflix
One thing to note is that the Itel P32 doesn't really have the ability to use the bandwidth that it nominally has. Looking at the top Google reviews, none of them mention this. The first review reads
Performance-wise, the phone doesn’t lag. It is powered by the latest Android 8.1 (GO Edition) ... we have 8GB+1GB ROM and RAM, to run on a power horse of 1.3GHz quad-core processor for easy multi-tasking ... I’m impressed with the features on the P32, especially because of the price. I would recommend it for those who are always on the move. And for those who take battery life in smartphones has their number one priority, then P32 is your best bet.
Itel mobile is one of the leading Africa distributors ranking 3rd on a continental scale ... the light operating system acted up to our expectations with no sluggish performance on a 1GB RAM device ... fairly fast processing speeds ... the Itel P32 smartphone delivers the best performance beyond its capabilities ... at a whooping UGX 330,000 price tag, the Itel P32 is one of those amazing low-range like smartphones that deserve a mid-range flag for amazing features embedded in a single package.
"Much More Than Just a Budget Entry-Level Smartphone ... Our full review after 2 weeks of usage ... While switching between apps, and browsing through heavy web pages, the performance was optimal. There were few lags when multiple apps were running in the background, while playing games. However, the overall performance is average for maximum phone users, and is best for average users [screenshot of game] Even though the game was skipping some frames, and automatically dropped graphical details it was much faster if no other app was running on the phone.
Notes on sites:
- Wix
- www.wix.com/website-template/view/html/3173?originUrl=https%3A%2F%2Fwww.wix.com%2Fwebsite%2Ftemplates%2Fhtml%2Fmost-popular&tpClick=view_button&esi=a30e7086-28db-4e2e-ba22-9d1ecfbb1250: this was the first entry when I clicked to get a theme
LCPwas misleading on every device- On the
Tecno Spark 8C, scrolling never really works. It's very jerky and this never settles down - On the
Itel P32, the page fails non-deterministically (different errors on different loads); it can take quite a while to error out; it was23son the first run, with the CPU pegged for28s
- Patreon
- www.patreon.com/danluu: used my profile where possible
- Scrolling on Patreon and finding old posts is so painful that I maintain my own index of my Patreon posts so that I can find my old posts without having to use Patreon. Although Patreon's numbers in the table don't look that bad in the table when you're on a fast laptop, that's just for the initial load. The performance as you scroll is bad enough that I don't think that, today, there exists a computer and internet connection that browse Patreon with decent performance.
- Threads
- threads.net/danluu.danluu: used my profile where possible
- On the
Itel P32, this technically doesn't load correctly and could be marked asFAIL, but it's close enough that I counted it. The thing that's incorrect is that profile photos have a square box around then- However, as with the other heavy pages, interacting with the page doesn't really work and the page is unusable, but this appears to be for the standard performance reasons and not because the page failed to render
- Twitter
- twitter.com/danluu: used my profile where possible
- Discourse
- meta.discourse.org: this is what turned up when I searched for an official forum.
- As discussed above, the
LCPis highly gamed and basically meaningless. We linked to a post where the Discourse folks note that, on slow loads, they put a giant splash screen up at2sto cap theLCPat2s. Also notable is that, on loads that are faster than the 2s, theLCPis also highly gamed. For example, on theM3 Maxwith low-latency1Gbpsinternet, theLCPwas reported as115ms, but the page loads actual content at1.1s. This appears to use the same fundamental trick as "Discourse Splash", in that it paints a huge change onto the screen and then carefully loads smaller elements to avoid having the actual page content detected as theLCP. - On the
Tecno Spark 8C, scrolling is unpredictable and can jump too far, triggering loading from infinite scroll, which hangs the page for3s-10s. Also, the entire browser sometimes crashes if you just let the browser sit on this page for a while. - On the
Itel P32, an error message is displayed after7.5s
- Bluesky
- bsky.app/profile/danluu.com
- Displays a blank screen on the
Itel P32
- Squarespace
- cedar-fluid-demo.squarespace.com: this was the second theme that showed up when I clicked themes to get a theme; the first was one called "Bogart", but that was basically a "coming soon" single page screen with no content, so I used the second theme instead of the first one.
- A lot of errors and warnings in the console with the
Itel P32, but the page appears to load and work, although interacting with it is fairly slow and painful LCPon theTecno Spark 8Cwas significantly before the page content actually loaded
- Tumblr
- www.tumblr.com/slatestarscratchpad: used this because I know this tumblr exists. I don't read a lot of tumblers (maybe three or four), and this one seemed like the closest thing to my blog that I know of on tumblr.
- This page fails on the
Itel P32, but doesn'tFAIL. The console shows that the JavaScript errors out, but the page still works fine (I tried scrolling, clicking links, etc., and these all worked), so you can actually go to the post you want and read it. The JS error appears to have made this page load much more quickly than it other would have and also made interacting with the page after it loaded fairly zippy.
- Shopify
- themes.shopify.com/themes/motion/styles/classic/preview?surface_detail=listing&surface_inter_position=1&surface_intra_position=1&surface_type=all: this was the first theme that showed up when I looked for themes
- On the first
M3/10run, Chrome dev tools reported a nonsensical697sof CPU time (the run completed in a normal amount of time, well under697sor even697/10s. This run was ignored when computing results. - On the
Itel P32, the page load never completes and it just shows a flashing cursor-like image, which is deliberately loaded by the theme. On devices that load properly, the flashing cursor image is immediately covered up by another image, but that never happens here. - I wondered if it wasn't fair to use this example theme because there's some stuff on the page that lets you switch theme styles, so I checked out actual uses of the theme (the page that advertises the theme lists users of the theme). I tried the first two listed real examples and they were both much slower than this demo page.
- Reddit
- reddit.com
- Has an unusually low
LCP*compared to how long it takes for the page to become usable. Although not measured in this test, I generally find the page slow and sort of unusable on Intel Macbooks which are, by historical standards, extremely fast computers (unless I use old.reddit.com)
- Mastodon
- mastodon.social/@danluu: used my profile where possible
- Fails to load on
Itel P32, just gives you a blank screen. Due to how long things generally take on theItel P32, it's not obvious for a while if the page is failing or if it's just slow
- Quora
- www.quora.com/Ever-felt-like-giving-up-on-your-dreams-How-did-you-come-out-of-it: I tried googling for quora + the username of a metafilter user who I've heard is now prolific on Quora. Rather than giving their profile page, Google returned this page, which appears to have nothing to do with the user I searched for. So, this isn't comparable to the social media profiles, but getting a random irrelevant Quora result from Google is how I tend to interact with Quora, so I guess this is representative of my Quora usage.
- On the
Itel P32, the page stops executing scripts at some point and doesn't fully load. This causes it to fail to display properly. Interacting with the page doesn't really work either.
- Substack
- Used thezvi.substack.com because I know Zvi has a substack and writes about similar topics.
- vBulletin:
- forum.vbulletin.com: this is what turned up when I searched for an official forum.
- Medium
- medium.com/swlh: I don't read anything on Medium, so I googled for programming blogs on Medium and this was the top hit. From looking at the theme, it doesn't appear to be unusually heavy or particularly customized for a Medium blog. Since it appears to be widely read and popular, it's more likely to be served from a CDN and than some of the other blogs here.
- On a run that wasn't a benchmark reference run, on the
Itel P32, I tried scrolling starting 35s after loading the page. The delay to scroll was5s-8sand scrolling moved an unpredictable amount, making the page completely unusable. This wasn't marked as aFAILin the table, but one could argue that this should be aFAILsince the page is unusable.
- Ghost
- source.ghost.io because this is the current default Ghost theme and it was the first example I found
- Wordpress
- 2024.wordpress.net because this is the current default wordpress theme and this was the first example of it I found
- XenForo
- xenforo.com/community/: this is what turned up when I searched for an official forum
- On the
Itel P32, the layout is badly wrong and page content overlaps itself. There's no reasonable way to interact with the element you want because of this, and reading the text requires reading text that's been overprinted multiple times.
- Wordpress (old)
- Used thezvi.wordpress.com because it has the same content as Zvi's substack, and happens to be on some old wordpress theme that used to be a very common choice
- phpBB
- www.phpbb.com/community/index.php: this is what turned up when I searched for an official forum.
- MyBB
- community.mybb.com: this is what turned up when I searched for an official forum.
- Site doesn't serve up a mobile version. In general, I find the desktop version of sites to be significantly better than the mobile version when on a slow device, so this works quite well, although they're likely penalized by Google for this.
- HN
- news.ycombinator.com
- In principle, HN should be the slowest social media site or link aggregator because it's written in a custom Lisp that isn't highly optimized and the code was originally written with brevity and cleverness in mind, which generally gives you fairly poor performance. However, that's only poor relative to what you'd get if you were writing high-performance code, which is not a relevant point of comparison here.
- danluu.com
- Self explanatory
- This currently uses a bit less CPU than HN, but I expect this to eventually use more CPU as the main page keeps growing. At the moment, this page has 176 links to 168 articles vs. HN's 199 links to 30 articles but, barring an untimely demise, this page should eventually have more links than HN.
- As noted above, I find that pagination for such small pages makes the browsing experience much worse on slow devices or with bad connections, so I don't want to "optimize" this by paginating it or, even worse, doing some kind of dynamic content loading on scroll.
- Woo Commerce
- I originally measured Woo Commerce as well but, unlike the pages and platforms tested above, I didn't find that being fast or slow on the initial load was necessarily representative of subsequent performance of other action, so this wasn't included in the table because having this in the table is sort of asking for a comparison against Shopify. In particular, while the "most default" Woo theme I could find was significantly faster than the "most default" Shopify theme on initial load on a slow device, performance was multidimensional enough that it was easy to find realistic scenarios where Shopify was faster than Woo and vice versa on a slow device, which is quite different from what I saw with newer blogging platforms like Substack and Medium compared to older platforms like Wordpress, or a modern forum like Discourse versus the older PHP-based forums. A real comparison of shopping sites that have carts, checkout flows, etc., would require a better understanding of real-world usage of these sites than I was going to get in a single day.
- NodeBB
- community.nodebb.org
- This wasn't in my original tests and I only tried this out because one of the founders of NodeBB suggested it, saying "I am interested in seeing whether @nodebb@fosstodon.org would fare better in your testing. We spent quite a bit of time over the years on making it wicked fast, and I personally feel it is a better representation of modern forum software than Discourse, at least on speed and initial payload."
- I didn't do the full set of tests because I don't keep the
Itel P32charged (the battery is in rough shape and discharges quite quickly once unplugged, so I'd have to wait quite a while to get it into a charged state) - On the tests I did, it got
0.3s/0.4son theM1and3.4s/7.2son theTecno Spark 8C. This is moderately slower than vBulletin and significantly slower than the faster php forums, but much faster than Discourse. If you need a "modern" forum for some reason and want to have your forum be usable by people who aren't, by global standards, rich, this seems like it could work. - Another notable thing, given that it's a "modern" site, is that interaction works fine after initial load, nothing crashed, etc.
- Sizes were
0.9 MB/2.2 MB, so also fairly light for a "modern" site and possibly usable on a slow connection, although that wasn't tested in this set of tests.
Another kind of testing would be to try to configure pages to look as similar as possible. I'd be interested in seeing that results for that if anyone does it, and maybe I'll try doing that on another day if no one else does it.
Appendix: this site vs. sites that don't work on slow devices or slow connections
Just as an aside, something I've found funny for a long time is that I get quite a bit of hate mail about the styling on this page (and a similar volume of appreciation mail). By hate mail, I don't mean polite suggestions to change things, I mean the equivalent of road rage, but for web browsing; web rage. I know people who run sites that are complex enough that they're unusable by a significant fraction of people in the world. How come people are so incensed about the styling of this site and, proportionally, basically don't care at all that the web is unusable for so many people?
Another funny thing here is that the people who appreciate the styling generally appreciate that the site doesn't override any kind of default styling, letting you make the width exactly what you want (by setting your window size how you want it) and it also doesn't override any kind of default styling you apply to sites. The people who are really insistent about this want everyone to have some width limit they prefer, some font they prefer, etc., but it's always framed in a way as if they don't want it, it's really for the benefit of people at large even though accommodating the preferences of the web ragers would directly oppose the preferences of people who prefer (just for example) to be able to adjust the text width by adjusting their window width.
Until I pointed this out tens of times, this iteration would usually start with web ragers telling me that "studies show" that narrower text width is objectively better, but on reading every study that exists on the topic that I could find, I didn't find this to be the case. Moreover, on asking for citations, it's clear that people saying this generally hadn't read any studies on this at all and would sometimes hastily send me a study that they did not seem to have read. When I'd point this out, people would then change their argument to how studies can't really describe the issue (odd that they'd cite studies in the first place), although one person cited a book to me (which I read and they, apparently, had not since it also didn't support their argument) and then move to how this is what everyone wants, even though that's clearly not the case, both from the comments I've gotten as well as the data I have from when I made the change.
Web ragers who have this line of reasoning generally can't seem to absorb the information that their preferences are not universal and will insist that they regardless of what people say they like, which I find fairly interesting. On the data, when I switched from Octopress styling (at the time, the most popular styling for programming bloggers) to the current styling, I got what appeared to be a causal increase in traffic and engagement, so it appears that not only do people who write me appreciation mail about the styling like the styling, the overall feeling of people who don't write to me appears to be that the site is fine and apparently more appealing than standard programmer blog styling. When I've noted this, people tend to become become further invested in the idea that their preferences are universal and that people who think they have other preferences are wrong and reply with total nonsense.
For me, two questions I'm curious about are why do people feel the need to fabricate evidence on this topic (referring to studies when they haven't read any, googling for studies and then linking to one that says the opposite of what they claim it says, presumably because they didn't really read it, etc.) in order to claim that there are "objective" reasons their preferences are universal or correct, and why are people so much more incensed by this than by the global accessibility problems caused by typical web design? On the latter, I suspect if you polled people with an abstract survey, they would rate global accessibility to be a larger problem, but by revealed preference both in terms of what people create as well as what irritates them enough to send hate mail, we can see that having fully-adjustable line width and not capping line width at their preferred length is important to do something about whereas global accessibility is not. As noted above, people who run sites that aren't accessible due to performance problems generally get little to no hate mail about this. And when I use a default Octopress install, I got zero hate mail about this. Fewer people read my site at the time, but my traffic volume hasn't increased by a huge amount since then and the amount of hate mail I get about my site design has gone from zero to a fair amount, an infinitely higher ratio than the increase in traffic.
To be clear, I certainly wouldn't claim that the design on this site is optimal. I just removed the CSS from the most popular blogging platform for programmers at the time because that CSS seemed objetively bad for people with low-end connections and, as a side effect, got more traffic and engagement overall, not just from locations where people tend to have lower end connections and devices. No doubt a designer who cares about users on low-end connections and devices could do better, but there's something quite odd about both the untruthfulness and the vitriol of comments on this.
- This estimate puts backwards-looking life expectancy in the low 60s; that paper also discusses other estimates in the mid 60s and discusses biases in the estimates. [return]
Arlington, VA
By Andrew Engelson On Friday, the state Senate Law and Justice Committee voted to pass Initiative 2113, which would repeal…
The post Legislature Moves Forward on Initiative Rolling Back Restrictions on Police Pursuits appeared first on PubliCola.
Oh look, hedge fund managers ruining more than just business.
Arlington, VA

Enlarge / The HP Envy 6020e is one of the printers available for rent. (credit: HP)
{kind=link}
{kind=link}
HP launched a subscription service today that rents people a printer, allots them a specific amount of printed pages, and sends them ink for a monthly fee. HP is framing its service as a way to simplify printing for families and small businesses, but the deal also comes with monitoring and a years-long commitment.
Prices range from $6.99 per month for a plan that includes an HP Envy printer (the current model is the 6020e) and 20 printed pages. The priciest plan includes an HP OfficeJet Pro rental and 700 printed pages for $35.99 per month.
HP says it will provide subscribers with ink deliveries when they're running low and 24/7 support via phone or chat (although it's dubious how much you want to rely on HP support). Support doesn't include on or offsite repairs or part replacements. The subscription's terms of service (TOS) note that the service doesn't cover damage or failure caused by, unsurprisingly, "use of non-HP media supplies and other products" or if you use your printer more than what your plan calls for.
New depths
Arlington, VA
Next Page of Stories